Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the aetiologic agent of the coronavirus disease 2019 (COVID-19). Since the earliest days of the pandemic, there have been many reports of central and peripheral neurological disease associated with the infection. Estimates of the frequency of neurological symptoms range from 30% patients admitted with confirmed COVID-19 [1] to 85% in patients in ICU or with acute respiratory distress syndrome (ARDS) [2,3,4]. Acute neurological complications are not only common, but they increase the short- and long-term burden of COVID-19 illness. For example, encephalopathy was independently associated with higher mortality within 30 days of hospitalization [1], and increased incidence of neuropsychiatric disorders within the 6 months after a COVID-19 diagnosis [5], raising the concern whether it is also an important risk factor for the neurological manifestations of the multi-organ syndrome PACS. While initial descriptions [6] were suggestive but unable to confirm a direct causal link between infection and neurological illness [7], an emerging epidemiological literature has shown that COVID-19 infection does appear to drive at least some of the neurological manifestations in the acute and long-term phase of the disease.
What is less clear is how these occur. Although in vivo animal studies have demonstrated the neurotropism of SARS-CoV-2, human studies do not support viral invasion of the nervous system as a major contributor [8]. Instead, a wide range of indirect mechanisms, often involving “immune dysregulation”, may converge in neurological dysfunction or damage. The cytokine storm triggered by the SARS-CoV-2 drives the delayed COVID-19 severity and it is likely involved in many of its neurological complications. However, following early reports of disorders such as Guillain–Barre syndrome (GBS) in the context of COVID-19 infection, studies have pointed to the development of neuronal autoimmunity as a potential important mechanism. Autoimmune neurological disorders have been associated with preceding viral infections in the past. From GBS, the prototypic post-infectious autoimmune disease, to neurological complications of influenza, Zika, and herpes simplex virus type one (HSV-1) infection, the latter associated with subsequent development of N-methyl-D-aspartate receptor antibody (NMDAR-antibody) encephalitis, many cases of postinfectious autoimmune aetiologies have been documented. These disorders may be associated with antibodies directed against self-antigens or tissues, that are thought to induce pathology at the level of the neuronal synapse, neuromuscular junction or myelin sheath [9,10,11,12]. In the context of COVID-19, however, the attribution of causality may be particularly difficult, requiring demonstration of two causally relevant processes (primary infection and secondary autoimmunity), often in the face of limited clinical and paraclinical information and overstretched healthcare resources. Determination of an immune-mediated aetiology is, however, crucial since it will likely mandate a distinct, frequently immunosuppressive, treatment approach. In this study we aimed to review the published cases of neuroimmune diseases described in association with COVID-19, assess the strength of this association, fulfilment of neurologic diagnostic criteria as well as clinical specificities, in order to provide guidance for the diagnosis and management of these emerging conditions.
Methods
We aimed at the following : (1) define the clinical and paraclinical characteristics and management of immune-mediated neurological complications of COVID-19. For that we performed a systematic review of the literature using a regularly updated Neurology and Neuropsychiatry of COVID-19 blog (https://blogs.bmj.com/jnnp/2020/05/01/the-neurology-and-neuropsychiatry-of-covid-19/) as source; and (2) describe the available evidence on the management of patients with pre-existing neuro-immunological disorders in the COVID-19 setting. Participants of the study were probable or confirmed cases of SARS-CoV-2 infection (WHO criteria: WHO/2019-nCoV/Surveillance_Case_Definition/2020.2) who developed immune-mediated neurological complications, that were further categorized using internationally accepted diagnostic criteria [13,14,15]. Only studies prior to 26/04/2021 with ≥ 5 participants were included. In addition, studies describing antibody-positive cases were included irrespective of the number of patients considering their relevance in understanding the mechanisms of these disorders. Complete details of the methods are available in the Supplementary Material.
Results
Pathogenesis of neuroimmune disorders
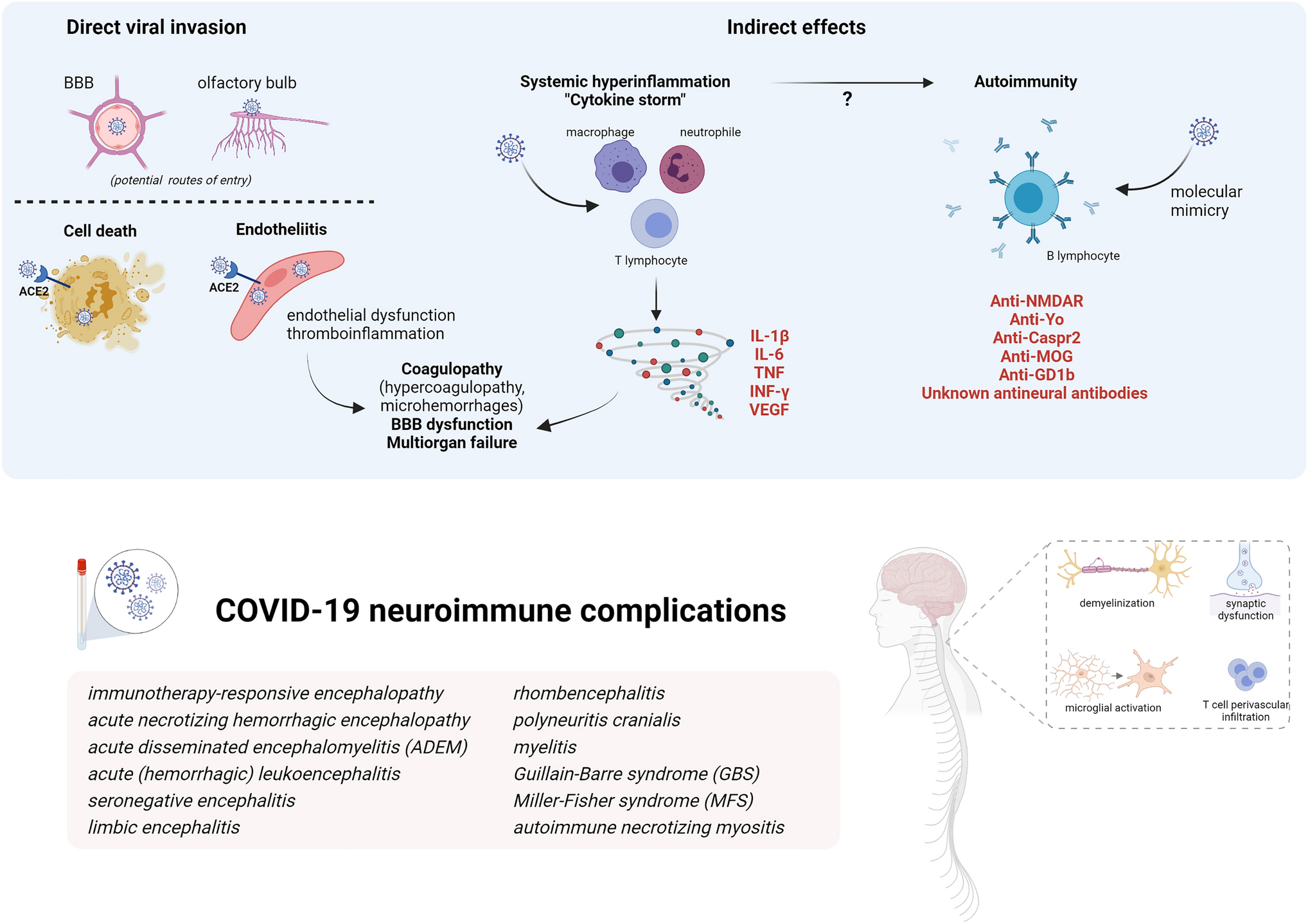
Mechanisms of SARS-CoV-2 pathogenesis in the nervous system are diverse (Fig.
1) and include direct or indirect effects. These mechanisms are not mutually exclusive and might act synergically.
Mechanisms of SARS-CoV-2 pathogenicity and immune-mediated effects on nervous system (Created with BioRender.com)
Potential routes of entries in the CNS for a direct viral invasion are by retrograde axonal transport from the olfactory system, by crossing the BBB or carried by infected immune cells. SARS-CoV-2 spike protein binds to angiotensin converting enzyme-2 (ACE2) for internalization, although other surface proteins may function as a co-factor. ACE2, a surface protein of many cell types, is highly expressed in the choroid plexus and found in neurons and astrocytes, oligodendrocytes, and in endothelial cells. Direct invasion of the virus can result in cell death or inflammatory infiltration of activated neutrophils and macrophages when invading endothelial cells (endotheliitis) which ultimately result in endothelial cell damage and thromboinflammation. Necropsy studies found activation of microglia and infiltration of cytotoxic T lymphocytes in brain parenchyma in some COVID-19 patients, suggestive of immune-mediated encephalitis. Among indirect effects, it has been demonstrated that SARS-CoV-2 is a potent trigger of innate and adaptive immune activation leading to overproduction of inflammatory cytokines, soluble mediators, hyperinflammation and multiorgan failure. Serum cytokine levels that are elevated in patients with COVID-19-associated cytokine storm include interleukin-1β, interleukin-6, IP-10, TNF, interferon-γ, macrophage inflammatory protein (MIP) 1α and 1β, and VEGF. This cytokine release syndrome may contribute to many of the clinical and laboratory findings reported in severe COVID-19: cytopenias, coagulopathy, hyperferritinemia and other acute-phase reactants (e.g., CRP, D-dimer) increase, endothelial damage and vascular permeability. In the brain, these cytokines can compromise the BBB and trigger a local amplification by inducing an innate immune response in resident cells which express toll-like receptors. Regarding autoimmunity, several antibodies against neuronal, glial or extraneural tissue are increasingly being described in both acute and recover in COVID-19 cases. The list of neuronal antibodies in the figure correspond to those exceptional COVID-19 cases described. It has been hypothesized that cross-reactivity due to molecular mimic could be the mechanism triggering this autoimmunity based on the molecular system involving gangliosides used by SARS-CoV2 to interact with the host cells and the detection of antiganglioside antibodies in cases of GBS after COVID-19.
Viral invasion
Direct effects consist of the SARS-CoV-2 nervous system invasion [8]. Although there is in vitro evidence of direct nervous system invasion and subsequent neuronal death, human neuropathological data have not yielded conclusive evidence of this mechanism so far and the most frequent histopathological findings are brain oedema, microthrombi, fresh ischaemic lesions and intense astrogliosis [8, 16]. Endothelial cell invasion and inflammatory infiltration, conversely, might be more relevant in pulmonary and extrapulmonary manifestations of COVID-19 [17]. Necropsy findings compatible with encephalitis do not correlate with the presence of virus in the brain; instead, they resemble the findings of immune-mediated encephalitis, predominantly in the brainstem and cerebellum [16].
Cytokine storm and autoimmunity
A major mechanism responsible for COVID-19 related severity is the ability to induce a systemic inflammation and cytokine storm following the initial replicative state [8, 18]. SARS-CoV-2 is a potent trigger of this immune hyperactivation which was initially described for certain systemic infections and well characterized for chimeric antigen receptor (CAR) T-cell therapy. High levels of cytokines can be detected in COVID-19 patients, both in serum and CSF, with worse prognostic associations and multiorgan failure. In particular, IL-6 might be a promising biomarker for severity and therapeutic decision-making, as antagonizing the IL-6 directly or through the JAK-STAT pathway has demonstrated improved prognosis in hospitalised COVID-19 patients with hypoxia and systemic inflammation [19, 20]. This cytokine storm has some specific features for COVID-19, for example it is frequently accompanied by lymphopenia in contrast to other disorders.
It is likely that the cytokine storm increases permeability of the blood brain barrier to potentially pathogenic circulating proteins (e.g. antibodies, other mediators) or to allow the systemic immune system to react aggressively against otherwise protected CNS antigens; unfortunately, data on blood brain barrier function is seldom available. It is also unknown whether the immune dysregulation associated with the cytokine storm contributes to some extent to activation and proliferation of autoreactive T cells initiating autoimmunity, the core pathophysiological mechanism underlying postinfectious disorders. Indeed, on the humoral arm of the immune system, multiple cases of potentially pathogenic CNS-targeting antibody responses have been described in association with both known and as yet uncharacterized neuronal or glial reactivities in COVID-19 patients. In one study, neutralizing human anti-SARS-CoV-2 antibodies showed cross-reactivity to unfixed murine tissue including brain, smooth-muscle, heart, lungs, kidney and colon [9]. Another clinical study found unexpectedly high rates of neuronal and glial antibodies in 11 patients presenting with varied neurological manifestations alongside COVID-19 infection [10]. Furthermore, potentially pathogenic antibodies to non-neuronal antigens, including antiphospholipid antibodies [11] are increasingly being described in both acute and recovered COVID-19 cases and have been variously associated with the severity of disease and outcome [12].
However, attribution of causality to self-reactive antibodies in patients with possible autoimmune neurological disease is far from straightforward, and even more challenging within a para-/postinfectious context. Evidence of pathogenicity for those antibodies against unknown nervous antigens has yet to be proven, and replication of these findings by other reference centres is awaited. Indeed, most studies of biomarkers in CSF from patients with CNS neurological conditions show a pattern more suggestive of cytokine release syndrome than antibody-mediated encephalitis, with increased levels of soluble mediators produced by the innate immune system (IL-6, TNF-α) and glial markers (GFAP), but with absence of intrathecal IgG synthesis and normal levels of chemokines associated with B/T-cell recruitment (CXCL13) [21,22,23]. Nevertheless, there is a recent interesting study analysing CSF and blood from individuals with COVID-19 with neurological symptoms, which found compartmentalized, CNS-specific T cell activation and B cell responses including antineuronal reactivity supporting autoimmunity in neurological complications [24].
Central nervous system (CNS) immune-mediated disorders
Due to different operational diagnostic definitions and the limitations of comprehensive studies during the pandemic, the incidence of CNS immune-mediated disorders remains unknown. Nevertheless, the awareness of these neuroimmune complications has progressively grown during the pandemic. A systemic review published in July 2020 reported 8 cases of encephalitis among 901 (0.9%) COVID19 patients with neurological manifestations [25]. Conversely, in a recently published surveillance study of acute neurological and psychiatric complications of COVID-19 across the UK including 267 cases, 25 (9.4%) corresponded to inflammatory CNS disorders [26]. To date, however, fewer than 200 cases of immune-mediated CNS cases have been described. In this section, we analyse series of 5 or more patients [27,28,29,30,31,32,33]. These 64 patients are individually reported in Table S1, and compared to peripheral syndromes in Table
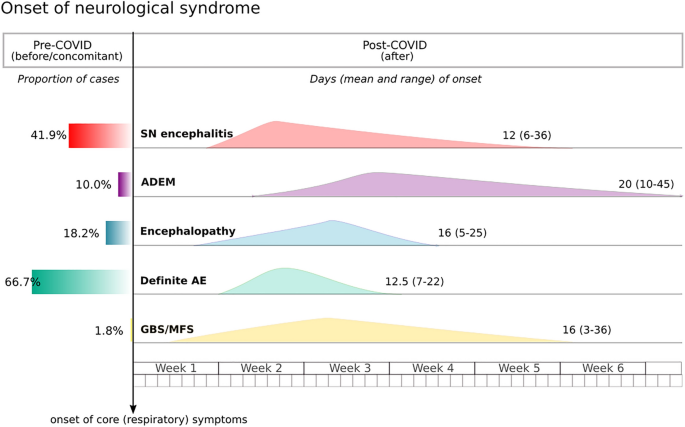
1. Figure
2 shows the temporal frame of neurological presentation in the COVID-19 evolution.
https://link.springer.com/article/10.1007/s00415-022-11050-w/tables/1
This graph shows the presentation of neurological symptoms across the different syndromic groups in reference to major COVID-19 symptoms (namely respiratory and fever). The left panel represents the proportion of patients presenting neurological symptoms before or concomitant to COVID-19 disease onset.
SN seronegative encephalitis,
AE autoimmune encephalitis,
ADEM acute disseminated encephalomyelitis,
GBS Guillain–Barré syndrome,
MFS Miller–Fisher syndrome.
Seronegative encephalitis
These cases had brain imaging or other evidence consistent with immune-mediated encephalitis and, apart from 9 patients diagnosed with ADEM described below, 43 patients (33% of total cohort; 17 females, mean [range 22–77] age of 60 years) met the criteria for autoimmune encephalitis based on clinical and paraclinical (MRI, CSF, EEG) criteria internationally accepted [13]. All of them were confirmed cases of COVID-19, and the presence of SARS-CoV-2 in the CSF was negative in all 38 that were tested. The neurological disorder occurred after COVID-19 presentation in 25 patients, with an approximate mean latency of 12 days from onset (range 6–36), but in the remaining 18 patients 16 presented simultaneously with COVID onset and in 2 patients the neurological disorder preceded it by 3–5 days [28]. Most patients had COVID-19 severe disease, and 38 required mechanical ventilation.
The most frequent neurological presentation reported was an acute brain dysfunction as encephalopathy or delirium, in 11 evident as prolonged weaning after sedation withdrawal. This was frequently accompanied by pyramidal signs and, cerebellar ataxia or brainstem dysfunction. One patient developed opsoclonus-myoclonus on top of confusion and hallucinations and another one presented a GBS concomitant to the encephalopathy. Nevertheless, combinations of psychiatric symptoms, prominent memory problems or seizures frequently described in non-COVID-related autoimmune encephalitis were not found in these series. Indeed, seizures (clinically or electrically defined) were reported only in 7 patients. Moreover, only 30 had some evidence of brain inflammation (20 suggestive MRI with normal CSF, 5 CSF pleocytosis with normal MRI and 5 with both abnormal).
Neuroimaging in these patients presents a practical challenge and the results further support a distinct type of encephalitis. A wide range of lesions were identified involving several brain areas. Only 3 of 43 patients had limbic encephalitis based on MRI (2 bilateral, 1 accompanied by diencephalic lesions). Other described lesions were confluent subcortical FLAIR/T2 hyperintensities, focal cortical and subcortical diffusion restriction, white-matter enhancing lesions, microbleeds, necrotic hemorrhagic lesions, and leptomeningeal enhancement. Leptomeningeal enhancement accompanied by bilateral frontotemporal hypoperfusion was one of the first imaging findings reported in 8/11 of patients admitted because of COVID-19-associated ARDS and neurological symptoms [34], but parenchymal abnormalities have been demonstrated to be more frequent in subsequent series. These lesions are located mainly in periventricular areas and centrum semiovale, but abnormalities in brainstem, cerebellar peduncles, basal ganglia, and corpus callosum were also described. A frequent radiological diagnosis was acute (haemorrhagic) leukoencephalitis, suggestive of a severe form of demyelinating disease [35]. A component of post–hypoxic leukoencephalopathy cannot be ruled out when symmetric confluent abnormalities without focal lesions predominate [30]. There were no bilateral thalamic lesions, hence no patient met criteria for the classic acute necrotizing encephalopathy described in other viral respiratory infections [
14].
In contrast to the significant imaging findings of these patients, the CSF parameters were generally normal. Only 10/43 patients had pleocytosis, and 1 had unmatched oligoclonal bands. IL-6 was elevated in the CSF from 2 patients. Hyperproteinorrachia is slightly more frequent (60% in the ENCOVID Study)[28], but this is a nonspecific finding that can be present in metabolic entities such as diabetes mellitus. None of the 21/43 patients tested had neuronal autoantibodies when tested in serum or CSF. Other laboratory findings reported in 10 patients (either with critical or mild COVID19 disease) were high levels of peripheral inflammatory markers (CRP, ferritin, and/or D-dimer), in line with results described in a cytokine storm [18].
Eight patients showed spontaneous improvement and did not receive any specific treatment; 27 patients were treated with immunotherapy (11 steroids, 4 IVIG, 4 plasma exchange, 7 combinations of 2 of the above, 1 IVIG combined with Tocilizumab), after which 16 improved. Outcomes are not comparable since reported timepoints were very different (in some patients, a complete neurological recovery has been described, whereas in others ICU discharge is the reported outcome). Seven patients (16%) died, only one among those with normal MRI. Complete recovery was reported in only two patients.