ME/CFS Science Blog
Senior Member (Voting Rights)
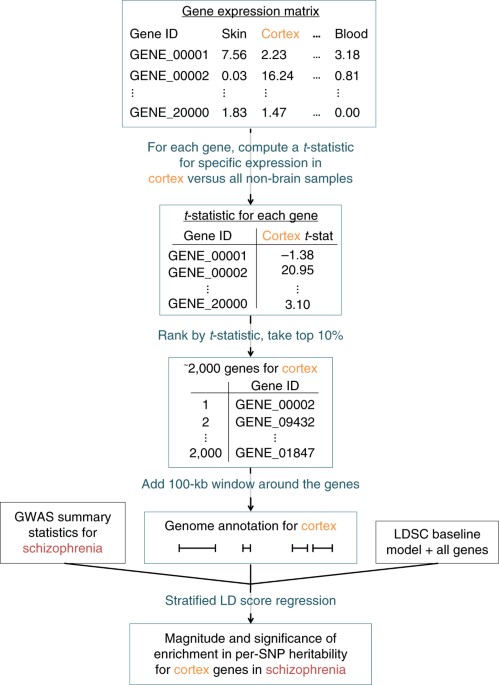
I thought it would be useful to make an overview of RNA datasets that we can match to the DecodeME DNA results. This could give us clues to which tissues or cell types are potentially involved in ME/CFS. I've made an overview of datasets below and plan to update it as we find new ones.
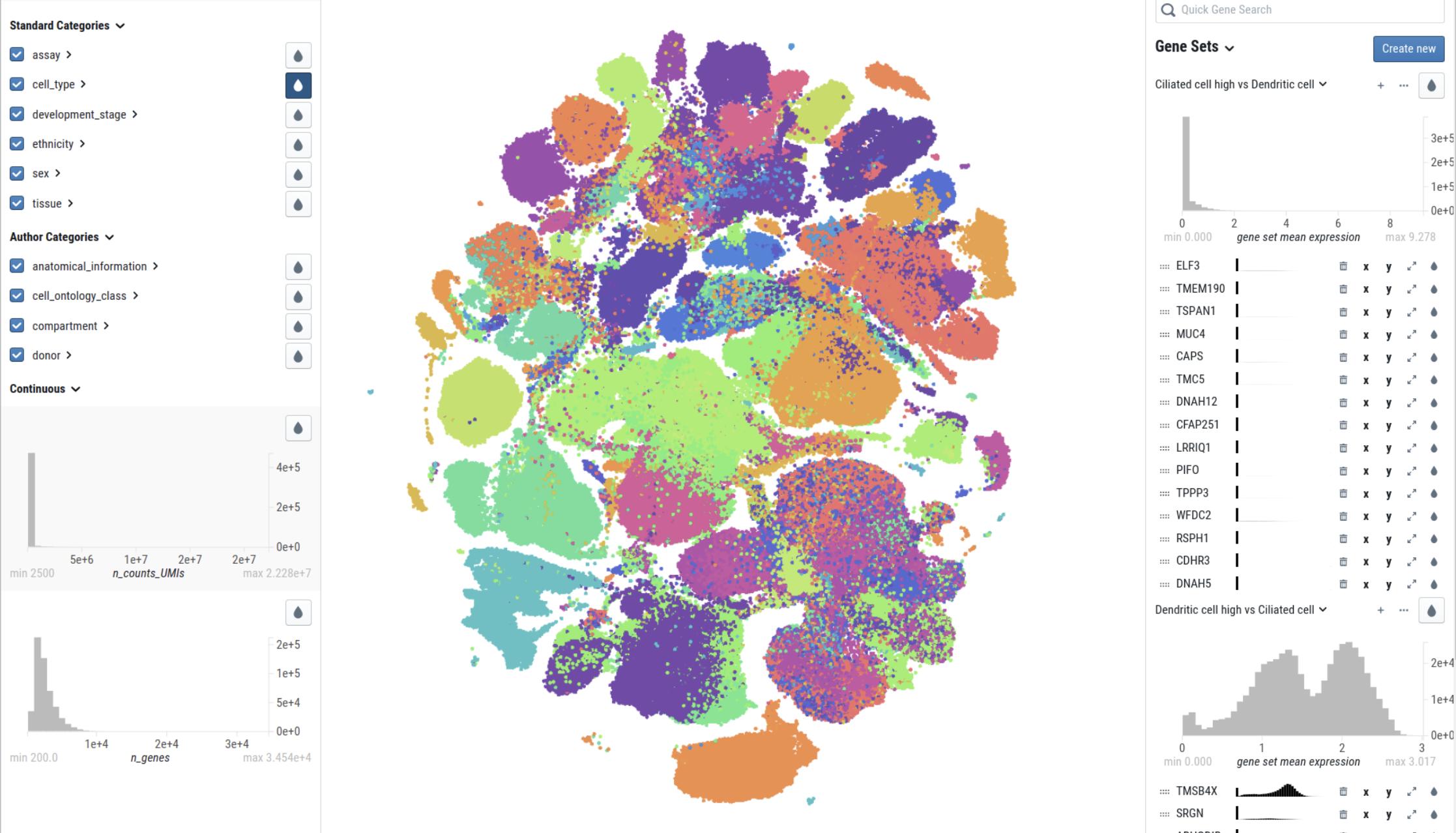
The Chan Zuckerberg Initiative’s CellxGene application is a nice respository where researchers can upload their data.
https://cellxgene.cziscience.com/
In the posts below, I'll share the results ME/CFS analyses that have been done with these datasets with a link to the relevant thread for more info. The current thread is only meant to provide an overview of all these analyses and discuss RNA datasets in general (in-depth discussion for a particular dataset or results are best reserved for the thread with the analysis itself).
The Chan Zuckerberg Initiative’s CellxGene application is a nice respository where researchers can upload their data.
https://cellxgene.cziscience.com/
In the posts below, I'll share the results ME/CFS analyses that have been done with these datasets with a link to the relevant thread for more info. The current thread is only meant to provide an overview of all these analyses and discuss RNA datasets in general (in-depth discussion for a particular dataset or results are best reserved for the thread with the analysis itself).
| Name | Animal | location | Population | Source |
| Genotype-Tissue Expression (GTEx) The GTEx Consortium, Science 2020. NIH funded. | Human | Entire body | Bulk-RNA 54 tissues 17,382 samples 838 postmortem donors | FUMA: “Gene expression values are log2 transformed average RPKM per tissue type after winsorized at 50 based on GTEx RNA-seq data. Tissue expression analysis is performed for 30 general tissue types and 53 specific tissue types separately. MAGMA was performed using the result of gene analysis (gene-based P-value) and tested for one side (greater) with conditioning on average expression across all tissue types.” https://fuma.ctglab.nl/tutorial#snp2gene |
| Human Brain Cell Atlas v1.0 Siletti et al. 2023 Linnarsson lab Karolinska Institute | Human | Brain | 3 adult donors ~3 million cells 31 superclusters, 461 clusters | https://cellxgene.cziscience.com/collections/283d65eb-dd53-496d-adb7-7570c7caa443 |
| DESCARTES Cao et al. 2020 Shendure lab Allen Institute | Human Fetus | Entire body | >110 fetal samples 15 organs ~4 million cells | https://cellxgene.cziscience.com/collections/c114c20f-1ef4-49a5-9c2e-d965787fb90c |
| Braun et al. 2023 Linnarsson lab Karolinska Institute | Human Fetus | Brain | 26 brain specimens 111 distinct biological samples First semester (5 – 14 weeks post-conception) ~ 1.6 million cells | https://cellxgene.cziscience.com/collections/4d8fed08-2d6d-4692-b5ea-464f1d072077 |
| Seeker 2023 CZI Cell Science | Human | Brain | White matter ~ 0.048 million cells | https://cellxgene.cziscience.com/collections/9d63fcf1-5ca0-4006-8d8f-872f3327dbe9 |
| Herring et al. 2022 (GSE168408) Lister lab | Human | Prefrontal Cortex | 26 postmortem samples spanning 6 developmental stages: Fetal, Neonatal, Infancy, Childhood, Adolescence, and Adult. ~0.15 million cells | https://brain.listerlab.org/ https://console.cloud.google.com/storage/browser/neuro-dev/Processed_data;tab=objects?prefix=&forceOnObjectsSortingFiltering=false |
| Allen Brain Atlas Sunkin et al. 2012 Allen Institute | Human | Brain | Two brains (older dataset) | https://atlas.brain-map.org/ https://human.brain-map.org/static/download |
| Franke lab data Fehrmann et al. 2015 | Human, mouse, rat | Entire body | Affymetrix methodology 33 tissues 77,840 samples GPL96: Homo sapiens: 17,309 GPL570: Homo sapiens: 37,427 GPL1261: Mus musculus: 17,081 GPL1355: Rattus norvegicus: 6,032 | Used by the DEPICT tool and the Finucane et al. 2018 paper. Is only available on a Google Cloud bucket where you have to pay for download costs. https://console.cloud.google.com/storage/browser/broad-alkesgroup-public-requester-pays/LDSCORE/LDSC_SEG_ldscores;tab=objects |
| Saunders et al. 2018 Dropviz Harvard | Mouse | Brain | ~0.69 million cells | http://dropviz.org/ |
| Mouse Brain Atlas Zeisel et al. 2015-2018 Linnarsson lab Karolinska Institute | Mouse | Brain | 9,970 cells 24 level 1 and 149 level 2 cell types Cortex, Hippocampus Hypothalamus Midbrain, Oligodendrocytes, Striatum | https://github.com/NathanSkene/MAGMA_Celltyping Used by Skene 2018, Olislagers 2018 |
| ImmGen | Mouse | 292 immune cell types | Array-based methodology Older version used in Finucane 2018: phase 1 (GSE15907) and phase 2 (GSE37448) data. | https://www.immgen.org/Databrowser19/DatabrowserPage.html Older version by Finucane 2018, available in Google Icloud bucket. |
| Chiou et al. 2023 Allen Institute BRAIN Initiative | Rhesus macaque | Brain | 5 animals 30 brain regions ~2.58 million cells | https://cellxgene.cziscience.com/collections/8c4bcf0d-b4df-45c7-888c-74fb0013e9e7 |
| Zhang et al. 2023 Allen Institute BRAIN Initiative | Mouse | Brain | 4 mice ~8.4 million cells | https://cellxgene.cziscience.com/collections/0cca8620-8dee-45d0-aef5-23f032a5cf09 |
Last edited: